De Canavan sjukdom Det är en sällsynt genetisk sjukdom som uppstår på grund av att nervceller i hjärnan är skadade och inte kan kommunicera med varandra. Denna sjukdom förekommer i alla samhällen och etniska grupper, även om den är mycket vanligare i den judiska befolkningen i Ashkenazi och deras ättlingar, där 1 av 6,400-13,000 människor drabbas. Den globala prevalensen är okänd.
Denna sjukdom ingår i gruppen av leukodystrofi. Denna kategori inkluderar alla genetiska störningar där myelinhöljet som omger nervcellernas axlar skadas och därför inte finns någon god kommunikation mellan nervceller..
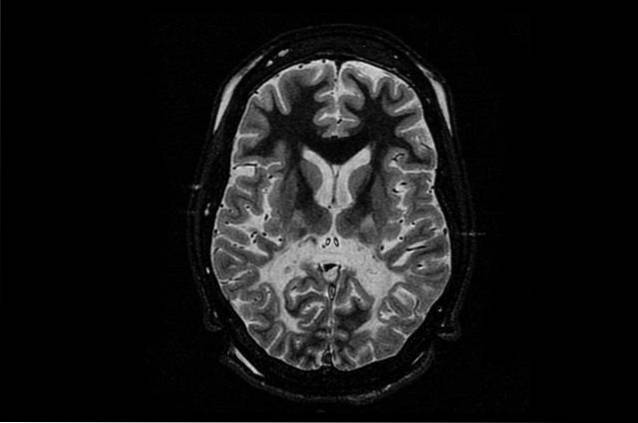
Den vanligaste och samtidigt mest allvarliga formen av denna sjukdom är neonatal eller infantil. Denna form av Canavan-sjukdom drabbar nyfödda barn eller under deras första levnadsår..
Barn som lider av denna sjukdom uppvisar inga problem under de första månaderna av livet, men dessa börjar blomstra mellan 3 och 5 månader. De huvudsakliga symtomen beror på utvecklingsbristen, där barn har motoriska problem som hindrar dem från att vända, vrida huvudet eller sitta utan stöd..
Andra vanliga symtom är muskelsvaghet (hypotoni), onormal huvudutveckling (makrocefali) och irritabilitet. I mindre utsträckning kan de också ha problem med att äta, anfall och sömnproblem..
En annan mindre vanlig form är Canavan-sjukdomen som börjar i mitten av barndomen eller tonåren. Barn och ungdomar med denna sjukdom har problem med språkutveckling och motoriska färdigheter, men dessa problem är ofta så milda att de inte identifieras som symtom på Canavans sjukdom..
Den förväntade livslängden för personer med Canavan-sjukdomen är mycket heterogen och varierar särskilt beroende på tidpunkten för sjukdomens början..
Barn som lider av neonatal eller infantil form lever vanligtvis bara några år, även om vissa når tonåren och mycket få fram till vuxen ålder. Medan de som lider av den unga formen har en normal livslängd.
Artikelindex
Det finns två väl differentierade former av Canavan-sjukdom: den av nyfödda eller infantil debut och den av uppkomst i medelbarndomen eller tonåren..
Symtom på nyfödda eller Canavans sjukdom är mycket allvarliga, vanligtvis märks inte förrän 3-50 månader, och inkluderar makrocephali, förlust av motorisk kontroll av huvudet och utvecklingsunderskott. Utvecklingsunderskott blir tydligare när barnet blir äldre.
De allvarligaste symptomen är de som är relaterade till motoriska problem, eftersom barn inte kan sitta eller stå upp utan stöd, gå eller tala. När de blir äldre kan hypotoni leda till spasticitet.
Även om de har alla dessa motoriska problem kan de lära sig att interagera socialt, le, peka på föremål ...
Vissa barn lider också av optisk atrofi, vilket orsakar synproblem, även om de fortfarande kan identifiera objekt visuellt.
När symtomen växer blir de värre och orsakar sömnsvårigheter, kramper och utfodringsbesvär. Barnet blir helt beroende och behöver hjälp för att utföra alla uppgifter.
Livslängden för dessa barn är ganska kort, de flesta dör på några år, även om vissa lever fram till tonåren eller vuxenlivet.
Canavan-sjukdomen med början i mellersta barndomen eller tonåren är mildare än den tidigare. Symtom inkluderar vissa svårigheter i verbal och motorisk utveckling.
Även om de vanligtvis är så milda att de inte identifieras som symtom på Canavan-sjukdomen, diagnostiseras denna sjukdom vanligtvis efter urinanalys, eftersom en av markörerna är den höga koncentrationen av N-acetylasparaginsyra (NAA i urinen.
Denna sjukdom orsakas av en mutation i en gen som heter ASPA. Denna gen är den som kontrollerar enzymet aspartoacylas, som är ansvarigt för nedbrytande NAA-molekyler..
Mutationen av ASPA-genen får aspartoacylas att minska dess effektivitet, så det kommer inte att bryta ner tillräckligt med NAA-molekyler och det kommer att finnas en hög koncentration av detta ämne. Ju tidigare denna mutation inträffar, desto sämre effekter har den.
Även om NAA-molekylernas funktion inte är så väl förstådd verkar det som om de är inblandade i transporten av vattenmolekyler genom nervceller och, överskottet av detta ämne, förhindrar att ny myelin bildas och förstör den befintliga. Detta orsakar att förbindelserna mellan nervceller inte fungerar ordentligt och hjärnan inte kan utvecklas normalt..
Vidare kan denna sjukdom ärvas på ett autosomalt recessivt sätt. Så om varje medlem i paret är bärare av den patogena varianten av ASPA-genen och de bestämmer sig för att få ett barn, kommer de sannolikt att:
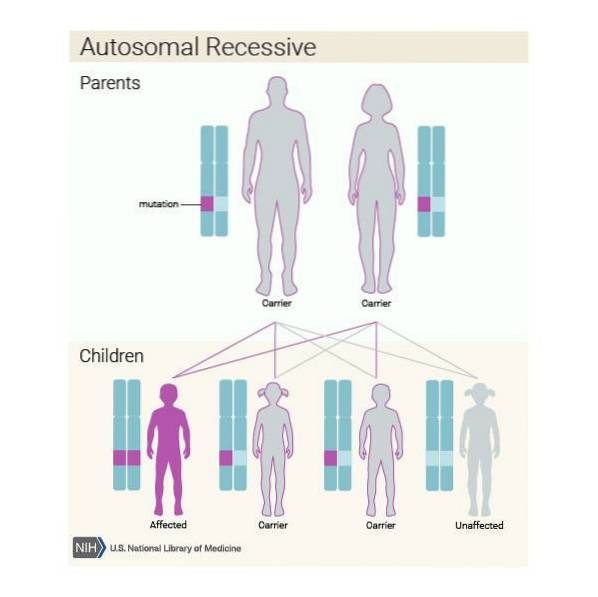
Det är mycket viktigt att individer som tillhör riskpopulationen, i detta fall ättlingar till Ashkenazi-judar, genomgår en genetisk analys för att kontrollera om de bär ASPA-genen innan de får barn..
Behandlingen beror på sjukdomsformen och de symtom som varje individ presenterar..
Det finns för närvarande inget botemedel mot Canavans sjukdom, så tillgängliga terapier fokuserar på att förbättra patientens livskvalitet genom att stödja, vårda och återfukta och förebygga och behandla infektioner.
Det rekommenderas att barn får sjukgymnastisk behandling för att förbättra sin hållning och motoriska färdigheter, för att undvika och behandla kontrakturer och muskelproblem, såsom trycksår. De kan också delta i terapeutiska och pedagogiska program för att förbättra sin kommunikationsförmåga..
Behandling med läkemedel inkluderar antiepileptiska läkemedel (AED) om barnet har anfall, acetazolamid (varumärke Diamox®) för att minska intrakraniellt tryck och injektioner av botulinumtoxin (Botox®) för att behandla spasticitet om den finns.
Det är nödvändigt att genomföra en uppföljning var sjätte månad för att kontrollera vilket tillstånd barnet befinner sig i och hur dess utveckling går.
Människor som lider av denna typ av sjukdom upplever mycket mildare symtom, så de behöver vanligtvis bara terapier för att förbättra sitt språk eller specialundervisningsprogram. De behöver ingen medicinering.
Årlig övervakning av barnets tillstånd rekommenderas.
Effekten av andra terapier studeras för närvarande i både människor och djurmodeller..
Effekten av en genetisk transplantation till hjärnan hos barn med Canavan-sjukdomen undersöks med en icke-viral vektor.
De första resultaten visar att denna typ av transplantation tolereras väl av barn och orsakar vissa biokemiska, radiologiska och metaboliska förändringar, men det är inte användbart för att bota sjukdomen, så test utförs fortfarande (Leone et al 2000, Janson et al. . till 2002).
McPhee et al. (2006) genomför en studie där den friska ASPA-genen transplanteras till olika ställen i barnens kropp med AAV2 som en vektor. I ett av testerna där 10 volontärbarn deltog. Hos tre av dem fungerade transplantationen och neutraliserade deras antikroppar, men ingen av barnen förbättrades.
Litiumcitrat kan minska nivån av NAA-koncentration i hjärnan, varför Assadi et al. (2010) beslutade att genomföra ett experiment där de administrerade litiumcitrat till 6 personer med Canavans sjukdom i 60 dagar.
NAA-koncentrationsnivåer i basala ganglier och vit substans i frontalloben hittades, även om inga kliniska förbättringar hittades.
Bristen på aspartoacylasenzymer orsakar låga nivåer av acetat i hjärnan, så Mahavarao och hans team (2009) bestämde sig för att ge glyceroltriacetat till två patienter med Canavals sjukdom för att höja sina acetatnivåer och se om det också ökade aspartoacylasnivåerna.
Föreningen tolererades väl av patienterna, även om inga kliniska förbättringar hittades. De genomför för närvarande tester som administrerar en högre mängd glyceroltriacetat.
Ett av sätten att skapa djurmodeller som representerar en sjukdom är att skapa djur knockout-. Dessa djur, vanligtvis möss, är genetiskt modifierade för att ta bort eller ändra genen som förändras i sjukdomen. I detta fall är den modifierade genen ASPA-genen..
Djurmodeller används för att bättre förstå sjukdomen, studera dess biologiska korrelat och verifiera effekten av nya behandlingar.
Matalon et al. (2003) använde möss knockout- för att testa effekten av en genterapi med AAV2 som en vektor. De fann att det hade skett förbättringar i myelinmantlarna, men bara i vissa delar, inte hela hjärnan.
Surendrans team i samarbete med Genzyme Corporation (2004) testade en stamcellstransplantationsbehandling. De fann att nya oligodendrocyter hade producerats, men inte tillräckligt för att återställa alla myelinmantlar..
Ett annat team testade en terapi som bestod av att ersätta de felaktiga asparthoacyklasenzymerna med nya som injicerades i bukhinnan hos mössen. knockout-.
De kortsiktiga resultaten visade att enzymerna kunde passera blod-hjärnbarriären (nådde sitt mål) och kunde signifikant minska nivåerna av NAA i hjärnan. Även om dessa resultat är lovande är en longitudinell studie nödvändig för att verifiera de långsiktiga effekterna (Zano et al., 2011).
De första tecknen som varnar läkare om att något är fel är de fysiska, särskilt hypotoni och makrocefali.
Om dessa tecken observeras utförs vanligtvis en neuroimaging-studie hos barnet för att kontrollera tecken på leukodystrofi, såsom en lägre densitet av vit substans. Det bör noteras att detta test är mindre effektivt hos barn med Canavan-sjukdom som börjar i mittenbarndomen eller tonåren..
När det väl har bevisats att barnet lider av en leukodystrofi görs mer specifika tester för att utesluta andra sjukdomar, dessa inkluderar:
Det sista steget för att bekräfta sjukdomen skulle vara att utföra en genetisk studie enligt följande:



Ingen har kommenterat den här artikeln än.