
De Aperts syndrom eller acrocephalosyndactyly typ I (ACS1) är en patologi av genetiskt ursprung som kännetecknas av närvaron av olika förändringar och missbildningar i skallen, ansiktet och extremiteterna.
På klinisk nivå kännetecknas Aperts syndrom av närvaron eller utvecklingen av en spetsig eller långsträckt skalle, nedsänkt ansiktsområde med en förändring i tändernas projektion, fusion och stängning av fingerben och leder, mental retardation variabel, språkstörningar , etc..
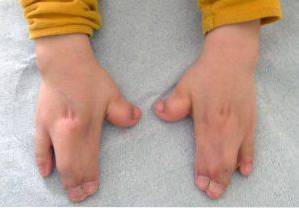
Även om denna patologi kan vara ärftlig, uppstår i de flesta fall Aperts syndrom utan närvaro av en familjehistoria, huvudsakligen på grund av en de novo-mutation under dräktighetsfasen..
De genetiska mekanismerna som orsakar Aperts syndrom är inte exakt kända. För närvarande har olika genetiska förändringar som kan producera denna patologi identifierats, huvudsakligen relaterade till mutationer i FGFR2-genen.
Å andra sidan börjar diagnosen av Aperts syndrom vanligtvis med klinisk misstanke under prenatalperioden efter identifiering av abnormiteter i rutinmässiga ultraljudsundersökningar och bekräftas genom en genetisk studie..
När det gäller behandling finns det ingen typ av botande ingrepp för Aperts syndrom. Under hela denna patologis historia har dock olika specifika ingrepp utformats som vanligtvis inkluderar neurokirurgi, kraniofacial kirurgi, maxillofacial kirurgi, läkemedelsbehandling, sjukgymnastik, psykologisk och neuropsykologisk intervention, bland andra..
Artikelindex
Apert syndrom är en genetisk patologi som kännetecknas av förekomsten av olika skelettmissbildningar på kranial-, ansikts- och / eller extremitetsnivå.
Den väsentliga förändringen av Aperts syndrom utgörs av en för tidig eller tidig tillslutning av kranialsprickorna, vilket orsakar en onormal tillväxt av resten av ansikts- och kranstrukturerna. Utöver dessa kan missbildningar också uppträda i övre och nedre extremiteterna, såsom fusion av fingrar och tår..
Å andra sidan kan de kognitiva förmågorna hos personer som lider av Aperts syndrom också påverkas, med varierande svårighetsgrad från mild till måttlig.
Även om Baumgartner (1842) och Wheaton (1894) nämnde de första om detta medicinska tillstånd, var det inte förrän 1906, när den franska medicinska specialisten Eugene Apert, noggrant beskrev detta syndrom och publicerade den första kliniska rapporten.
I sin publikation, Eugene Apert, beskriver en uppsättning nya fall av patienter som drabbats av ett väldefinierat missbildningsmönster och kännetecknas av de karaktäristiska tecknen och symptomen på denna patologi..
Det var således först 1995 som de etiologiska genetiska faktorerna för Aperts syndrom identifierades. Specifikt beskrev Wilkie et al. Närvaron av två mutationer i FGFR2-genen hos cirka 40 drabbade patienter..
Dessutom är Aperts syndrom ett medicinskt tillstånd som klassificeras inom de sjukdomar eller patologier som kännetecknas av kraniosynostos (för tidig tillslutning av kranial suturer).
Andra patologier som tillhör denna grupp är Pfeiffer syndrom, Crouzon syndrom, Saethre-Chotzcen syndrom och Carpenter syndrom..
Apert syndrom anses vara en sällsynt eller sällsynt patologi, det vill säga det har en prevalens på mindre än ett fall per 15 000 invånare i den allmänna befolkningen..
Specifikt förekommer Aperts syndrom omkring en person för varje 160 000-200 000 födda och dessutom finns det en 50% sannolikhet att överföra denna patologi på ärftlig nivå.
Dessutom har en högre prevalens hos män och kvinnor inte identifierats med avseende på fördelningen efter kön, och den har inte heller förknippats med särskilda etniska grupper eller geografiska platser..
För närvarande och med tanke på att Aperts syndrom identifierades ungefär 1984, i kliniska rapporter och i den medicinska litteraturen som har publicerat mer än 300 fall av denna patologi.
De kliniska manifestationerna av Apert syndrom inkluderar vanligtvis en missbildning eller ofullständig utveckling av kranialstrukturen, en atypisk fenotyp eller ansiktsmönster och skelettförändringar i extremiteterna..
När det gäller Aperts syndrom är det centrala engagemanget relaterat till bildandet och stängningen av benets struktur i skallen. Under embryonal utveckling inträffar en process som kallas kreneosynostos, som kännetecknas av för tidig stängning av kraniella suturer.
Kranifissurer eller suturer är en typ av fibrösa vävnadsband som har det grundläggande målet att förbinda benen som utgör skallen (frontal, occipital, parietal och temporal).
Under dräktighetsfasen och den tidiga postnatala perioden hålls benstrukturen som utgör skallen samman tack vare dessa fibrösa och elastiska vävnader.
Normalt smälter inte kranialbenen förrän omkring 12 till 18 månader. Förekomsten av mjuka fläckar eller mellanrum mellan kranialbenen är en del av normal barns utveckling.
Under hela barndomsstadiet tillåter dessa suturer eller flexibla regioner hjärnan att växa på ett påskyndat sätt och dessutom skydda den från stötar.
Således, i Aperts syndrom, gör för tidig tillslutning av dessa kraniala suturer och kranialben normal utveckling av kranial och hjärntillväxt omöjlig..
Följaktligen kan de vanligaste tecknen och symtomen på Aperts syndrom inkludera:
Dessa typer av förändringar påverkar främst de övre och nedre extremiteterna, normalt fusion och utveckling av fingrarna..
Förutom dessa är det också möjligt att observera andra kliniska fynd på muskuloskeletala nivån, förkortning av olika ben (radie, ben, lårben), hypoplasi i skulderbladet eller bäckenet, fusion av livmoderhalsen.
Som en följd av detta kommer många drabbade att uppvisa minskad ledrörlighet och kan därför utveckla olika svårigheter för förvärv av grov- och finmotorik.
Dessa typer av anomalier är mycket heterogena och varierande bland drabbade individer, men några av de vanligaste har identifierats:
Den etiologiska förändringen av denna patologi kan leda till utveckling av lesioner eller sekundära patologier på en morfologisk och strukturell nivå i olika delar av kroppen, några av dem inkluderar:
Trots det faktum att det i många fall är möjligt att observera förekomsten av en allmän förändring av kognitiva funktioner och intellektuell nivå, är mental retardering inte entydigt närvarande i alla fall av Aperts syndrom..
Dessutom, i fall där det finns en försämring av den intellektuella nivån, kan detta variera, på en skala från mild till måttlig.
Å andra sidan, inom det språkliga området, är utvecklingen av olika underskott frekvent, huvudsakligen relaterad till artikulation av ljud som ett resultat av underkäken och orala missbildningar..
Aperts syndrom beror på närvaron av en specifik mutation i FGFR2-genen. Experimentella studier har visat att denna gen är ansvarig för produktionen av ett protein, kallat fibroblasttillväxtfaktorreceptor 2.
Bland funktionerna för denna faktor beskriver den sändningen av olika kemiska signaler till omogna celler för att orsaka deras transformation och differentiering till benceller under fostrets eller prenatala utvecklingsfas..
Därför förändrar närvaron av mutationer i FGFR2-genen funktionen hos detta protein och kan därför orsaka en tidig fusion av benen i skallen, handen och fötterna..
En god del av de kliniska egenskaperna hos Aperts syndrom kan identifieras under graviditeten, särskilt vid ultraljudundersökningar av graviditet och fosterutveckling..
När det finns en klinisk misstanke startas alltså en genetisk studie för att identifiera närvaron av en genetisk mutation som är kompatibel med Aperts syndrom..
Å andra sidan, när tecknen är subtila eller inte har identifierats före födseln, efter detta är det möjligt att utföra en detaljerad fysisk analys och olika genetiska tester för att bekräfta diagnosen..
Även om det inte finns något specifikt botemedel mot Aperts syndrom, har olika metoder beskrivits för behandling av symtom och medicinska komplikationer av denna patologi..
De mest effektiva terapeutiska ingripandena är de som genomförs tidigt, i de första ögonblicken och involverar yrkesverksamma från olika områden.
Vanligtvis kräver behandling av drabbade barn individuell planering, med flera operationer planerade. Således är hanteringen av denna patologi baserad på korrigering av skelett- och kranio-ansiktsmissbildningar och psykologiskt och neuropsykologiskt stöd..
Genom neurokirurgi är målet att rekonstruera kranialvalvet, medan specialister inom maxillofacialkirurgi försöker korrigera missbildningar i ansiktet. Å andra sidan deltar också traumakirurger ofta för rekonstruktion av missbildningar i händer och fötter.
Dessutom är utformningen av individualiserade program för tidig stimulering, kommunikationsrehabilitering, träning av sociala färdigheter eller psykopedagogisk uppföljning fördelaktiga för att uppnå en optimal, funktionell och oberoende utveckling av de drabbade individerna..



Ingen har kommenterat den här artikeln än.