
De Williams syndrom det är en utvecklingsstörning av genetiskt ursprung som är förknippad med en karakteristisk profil för fysiska och kognitiva försämringar. Specifikt på klinisk nivå kännetecknas det av fyra kardinalpunkter: 1) atypiska ansiktsdrag och egenskaper, 2) generaliserad fördröjning av psykomotorisk utveckling och specifik neurokognitiv profil, 3) kardiovaskulära förändringar och t) möjlighet att utveckla infantil hyperkalcemi.
Trots att Williams syndrom anses vara en sällsynt sjukdom finns det tusentals drabbade människor runt om i världen. När det gäller diagnosen tillhandahåller den kliniska undersökningen vanligtvis de nödvändiga resultaten för dess upprättande, men för att utesluta andra patologier och falska positiva inleds vanligtvis en genetisk studie med olika tekniker.
Å andra sidan finns det varken ett botemedel mot Williams syndrom eller ett standardbehandlingsprotokoll, så de flesta terapeutiska ingripanden kommer att försöka reglera medicinska komplikationer. Dessutom kommer det att vara viktigt att inkludera tidiga vårdprogram, individualiserad specialundervisning och neuropsykologisk stimulering i interventionerna..
Artikelindex
Williams syndrom är en utvecklingsstörning som signifikant kan påverka olika områden.
Generellt kännetecknas denna patologi av närvaron av atypiska ansiktsdrag eller kardiovaskulära förändringar, måttlig intellektuell funktionsnedsättning, inlärningsproblem och distinkta personlighetsdrag..
Således beskrevs den första patienten med Williams syndrom av Dr. Guido Fanconi i en klinisk rapport från 1952. Det var emellertid kardiologen Joseph Williams som 1961 exakt identifierade denna patologi, såväl som den beskrivs av tyska Beuren..
På grund av detta får Williams syndrom sitt namn från båda författarna (Williams-Beuren syndrom), eller helt enkelt från den första.
Trots att identifieringen av patologin fram till för några år sedan utfördes baserat på de fenotypiska egenskaperna, fann Edward et al 1993. Genetisk abnormitet i kromosom 7q 11.23 som etiologisk orsak..
Även om Williams syndrom är förknippat med ett stort antal sekundära medicinska komplikationer, har det inte en hög dödlighet. I många fall kan drabbade individer uppnå en oberoende funktionell nivå.
Williams syndrom anses vara en sällsynt eller sällsynt genetisk störning.
Williams Syndrome Association, bland andra institutioner, har uppskattat att Williams syndrom har en prevalens på cirka 1 fall per 10 000 människor världen över. Specifikt har det identifierats att det i USA kan vara omkring 20 000 eller 30 000 drabbade.
När det gäller fördelningen av patologin efter kön finns det inga senaste data som indikerar en högre prevalens hos någon av dem. Dessutom har inga skillnader identifierats mellan geografiska regioner eller etniska grupper.
Å andra sidan vet vi också att Williams syndrom är ett sporadiskt medicinskt tillstånd, även om vissa fall av familjeöverföring har beskrivits.
Williams syndrom, liksom andra patologier av genetiskt ursprung, har en klinisk kurs som kännetecknas av multisysteminvolvering.
Många författare, som González Fernández och Uyaguari Quezada, beskriver det kliniska spektrumet av Williams syndrom kategoriserat i flera områden: biomedicinska egenskaper, psykomotoriska och kognitiva egenskaper, psykologiska och beteendemässiga egenskaper, bland andra..
Den fysiska påverkan som finns i Wiliams syndrom är olika, bland de vanligaste kliniska fynd vi kan observera:
Försenad eller långsam utveckling kan redan upptäckas under graviditeten. Barn som drabbas av Williams syndrom föds ofta med låg vikt och längd. Dessutom, när vuxenstadiet har uppnåtts, är den totala höjden vanligtvis lägre än den allmänna befolkningen, cirka 10-15 cm.
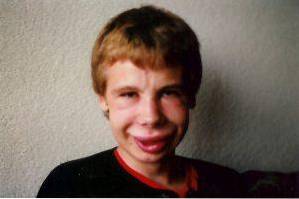
Ansiktsförändringar är en av de mest karakteristiska kliniska fynden i detta syndrom. Hos drabbade individer kan vi observera en signifikant smal panna, markerade hudveck i palpebral spricka, strabismus, stellat iris, kort och tillplattad näsa, framträdande kindben och en mindre haka än vanligt..
Vid förändringar relaterade till utveckling av muskler och ben är det möjligt att observera förekomsten av minskad muskeltonus och styrka, ledlöshet, skolios, kontrakturer, bland andra. Visuellt kan en hållning som kännetecknas av hängande axlar och halvböjda nedre extremiteter observeras..
Även om det vanligtvis inte finns några signifikanta avvikelser eller missbildningar i den auditiva pinnan, utvecklas i alla fall en ökning av hörselkänsligheten. Berörda individer tenderar att uppfatta eller uppleva vissa ljud som irriterande eller smärtsamma.
Huden har vanligtvis liten elasticitet, så det är möjligt att observera tidiga tecken på åldrande. Dessutom kan bråck utvecklas, särskilt i inguinal och navelregionen..
Olika abnormiteter i hjärtat och blodkärlen utgör den mest betydande medicinska komplikationen, eftersom de kan äventyra den drabbade personens överlevnad..
Bland de kardiovaskulära anomalierna är några av de vanligaste supravalvulära aortastenos, lunggrenstenos, aortaklaffstenos. Alla dessa förändringar, på klinisk nivå, kan påverka andra vaskulära territorier och till och med hjärnan på grund av utvecklingen av arteriell hypertoni.
Avvikelser relaterade till njurfunktion och urinblåsa är mycket vanliga. Dessutom kan en ackumulering av kalcium (nefrokalcinos), brådskande urinvägar eller nattlig enurese detekteras.
På kognitiv nivå utgörs de viktigaste egenskaperna av en generaliserad fördröjning av förvärvet av motoriska färdigheter, måttlig intellektuell fördröjning och olika förändringar relaterade till visuell perception.
Olika förändringar relaterade till balans- och koordinationsproblem beskrivs, vilka huvudsakligen beror på förekomsten av muskuloskeletala abnormiteter och som bland annat kommer att orsaka en fördröjning av gångförvärvet, slutliga motoriska färdigheter etc..
Det är möjligt att hitta en måttlig mental retardation, den typiska IQ för de drabbade svänger vanligtvis mellan 60 och 70. När det gäller de specifika områden som påverkas finns det en tydlig asymmetri: förutom psykomotorisk koordination, perception och visuell integration är det vanligtvis påverkas tydligt, medan områden som språk vanligtvis är mer utvecklade.
I de allra första inledningarna är det vanligtvis en fördröjning i förvärvet av språkkunskaper, men det återhämtar sig vanligtvis runt 3-4 år. Barn med Williams syndrom tenderar att ha bra uttrycksfull kommunikation, kan använda kontextualiserad vokabulär, korrekt grammatik, ögonkontakt, ansiktsuttryck, etc..
En av de viktigaste resultaten i Williams syndrom är det exceptionella sociala beteendet hos de drabbade. Även om ångestkriser eller överdrivna bekymmer i vissa fall kan uppstå är de mycket empatiska och känsliga.
Den senaste forskningen har visat att orsaken till Williams syndrom finns i olika genetiska förändringar på kromosom 7. Kromosomer bär varje persons genetiska information och ligger i kärnan i kroppens celler.
Hos människor kan vi hitta 46 kromosomer som fördelas parvis. Dessa är numrerade från 1 till 23, förutom det sista paret som består av könskromosomer, kallat XX för kvinnor, XY för män. Således kan det finnas en oändlighet av gener i varje kromosom.
Specifikt är den onormala processen som identifierats i Williams syndrom en mikroelektion eller nedbrytning av en DNA-molekyl som bekräftar denna kromosom. Normalt sker denna typ av fel i utvecklingsfasen för manliga eller kvinnliga könsceller..
Genetiska abnormiteter finns i området 7q11.23, där mer än 25 olika gener relaterade till det karaktäristiska kliniska mönstret för denna patologi har identifierats..
Några av generna, såsom Clip2, ELN, GTF21, GTF2IRD1 eller LIMK1, är frånvarande hos de drabbade. Förlust av ELN är relaterat till bindväv, hud och kardiovaskulära abnormiteter.
Å andra sidan visar viss forskning att förlusten av Clip2-, GTF2I-, GTF2IRD1- och LIMK1-generna kan förklara förändringar i visuoperceptiva processer, beteendefenotyp eller kognitiva underskott.
Dessutom verkar specifikt GTF2IRD1-genen spela en framträdande roll i utvecklingen av atypiska ansiktsdrag. För sin del verkar NCF1-genen vara relaterad till en hög risk att utveckla högt blodtryck.
Fram till de senaste åren ställdes diagnosen av Williams syndrom uteslutande baserat på observation av fenotypiska egenskaper (ansiktsförändringar, intellektuell funktionsnedsättning, specifika kognitiva underskott, bland andra).
För närvarande görs dock diagnosen Williams syndrom vanligtvis i två steg: analys av de kliniska fynden och bekräftande genetiska studier. Således innefattar den kliniska diagnosen vanligtvis:
- Fysisk och neurologisk undersökning och utvärdering.
- Analys av tillväxtparametrar.
- Undersökning av hjärt-andningsorganen.
- Nephrourological undersökning.
- Analys av kalciumnivåer i urin och blod.
- Oftalmologisk analys.
Å andra sidan används genetisk analys för att bekräfta närvaron av genetiska förändringar som är kompatibla med Williams syndrom, bland de vanligaste testerna är den fluorescerande in situ hybridiseringstekniken (FIHS)..
Efter extraktion av ett blodprov utförs hybridiseringstekniken in situ genom att markera DNA-sonder som detekteras under ett fluorescerande ljus..
Det finns ingen specifik behandling för Williams syndrom, men denna patologi är associerad med flera komplikationer i olika organ, så medicinska ingripanden kommer att vara inriktade på behandlingen av dessa.
Författarna González Fernández och Uyaguari Quezada betonar att alla ingripanden måste ha en tydlig tvärvetenskaplig karaktär, vilket möjliggör behandling av den symtomatiska variationen som är karakteristisk för detta syndrom. Dessutom påpekar de olika terapeutiska åtgärder beroende på det drabbade området:
I det här fallet kräver medicinska komplikationer såsom hjärtförändringar eller missbildningar i muskuloskeletala vanligtvis behandling baserad huvudsakligen på administrering av läkemedel och kirurgiska ingrepp. Läkare från olika områden (barnläkare, kardiologer, ögonläkare etc.) deltar vanligtvis i behandlingen av fysiska symtom..
Kognitiva brister som visuell-perceptuell förändring eller språklig fördröjning bör åtgärdas från de tidiga stadierna. Kognitiv stimulering och rehabilitering kommer att vara en avgörande faktor för att uppnå ett autonomt liv under vuxenlivet.
Även om de som drabbas av Williams syndrom vanligtvis har god social funktion, tenderar de vid vissa tillfällen att visa alltför oroliga beteenden och utveckla uthålligt beteende eller fobier.
Därför är det i dessa fall viktigt att genomföra en psykologisk strategi genom olika strategier som är effektiva för att minimera dessa problem eller svårigheter..


Ingen har kommenterat den här artikeln än.