
De Prader-Willis syndrom (SPW) är en multisystemisk patologi som har ett medfödd genetiskt ursprung. Det är en komplex sjukdom som påverkar aptit, tillväxt, ämnesomsättning, beteende och / eller kognitiv funktion.
På klinisk nivå, under barndomsstadiet, kännetecknas denna sjukdom av förekomsten av olika medicinska fynd som muskelsvaghet, ätstörningar eller generaliserad utvecklingsfördröjning..
Dessutom, på kognitiv och beteendemässig nivå, uppvisar en god del av individerna som påverkas av Prader-Willis syndrom en måttlig intellektuell försämring eller fördröjning som åtföljs av olika inlärnings- och beteendeproblem.
Även om Prader-Willis syndrom anses vara en sällsynt eller ovanlig sjukdom, indikerar många studier att det är en av de vanligaste patologierna i det genetiska området. Diagnosen av denna sjukdom görs huvudsakligen på grundval av kliniska resultat och kompletterande genetiska tester..
När det gäller behandling har ett botemedel mot Prader-Willis syndrom ännu inte identifierats, så det terapeutiska tillvägagångssättet är inriktat på behandling av symtom och komplikationer, med fetma som det medicinska fynd som utgör det största hotet för de drabbade.
Således, i förhållande till prognosen och livskvaliteten, kommer båda att bero på svårighetsgraden av de associerade medicinska problemen och de beteendemässiga eller kognitiva störningar som kan utvecklas.
Artikelindex
Olika kliniska rapporter tyder på att Prader-Willis syndrom (PWS) ursprungligen beskrevs av J. L. Down 1887, efter att ha diagnostiserat en av hans patienter med ”polysarcia”..
Det var dock Drs Prader, Labhart och Willi som 1956 beskrev ytterligare 9 fall och gav denna patologi sitt namn. Dessutom systematiserades egenskaperna och diagnostiska kriterierna för Prader-Willis syndrom av Holm et al..
Prader-Willis syndrom är en medfödd genetisk förändring, det vill säga det är en patologi som är närvarande från födseln och kommer att påverka individen under hela sitt liv om det inte finns något botande terapeutiskt ingrepp..
Denna patologi presenterar en komplex klinisk kurs som kännetecknas av många medicinska manifestationer.
Även om fenotypen av Prader-Willis syndrom idag är mer känd, har det varit de senaste 25 åren när det har gjorts betydande framsteg i analysen och förståelsen av denna sjukdom..
Uttrycket av Prader-Willis syndrom är olika, det tenderar att påverka flera system och strukturer, de flesta förändringarna är relaterade till hypotalamus dysfunktion..
Hypotalamus är en neurologisk struktur som har en väsentlig roll i kontrollen av homeostatiska funktioner: reglering av hunger, törst, sömnväckningscykler eller reglering av kroppstemperatur..
Dessutom frigör hypotalamus olika hormoner till olika körtlar: tillväxt, sexuell, sköldkörtel etc..
Slutligen måste vi påpeka att Prader-Willis syndrom också kan visas i den medicinska och experimentella litteraturen med andra termer som Prader-Labhart-Willis syndrom eller med akronymen PWS..
Andra synonymer är också Labhart Willi syndrom, Praser Labhart Willi Fancone syndrom eller hypogenital dystrofi syndrom..
Prader-Willis syndrom (PWS) är en sällsynt genetisk sjukdom. Termen sällsynt sjukdom (ER) används för att hänvisa till de patologier som är sällsynta eller få personer som lider av den.
För närvarande uppskattas det att Prader-Willis syndrom är en patologi med en ungefärlig frekvens på 1 fall per 10 000–30 000 människor världen över.
Å andra sidan, när det gäller fördelningen efter kön, har det observerats att denna patologi drabbar män och kvinnor lika, och den är inte associerad med etniska grupper eller geografiska regioner..
Dessutom anses Prader-Willis syndrom vara den främsta orsaken till fetma av genetiskt ursprung.
På klinisk nivå har Prader-Willis syndrom traditionellt associerats med neonatal hypotoni, hypogonadism, hyperfagi, fetma, kort kroppsvikt, generaliserad fördröjning i utvecklingen, måttlig intellektuell funktionsnedsättning, atypiskt ansiktsutseende och olika beteendeförändringar..
Trots detta är det kliniska uttrycket för denna patologi mycket heterogent och varierar signifikant mellan drabbade individer..
Dessutom tenderar de karakteristiska tecknen och symtomen på Prader-Willis syndrom att variera med biologisk utveckling, så vi kan observera olika kliniska fynd i fostrets och nyfödda perioden, spädbarnsperioden eller tidig barndom, skolstadiet och slutligen scenen Teen.
På ett systematiskt sätt beskriver José A. del Barrio del Campo och medarbetare i detalj de mest karakteristiska förändringarna inom det biomedicinska, psykomotoriska, kognitiva och beteendemässiga området:
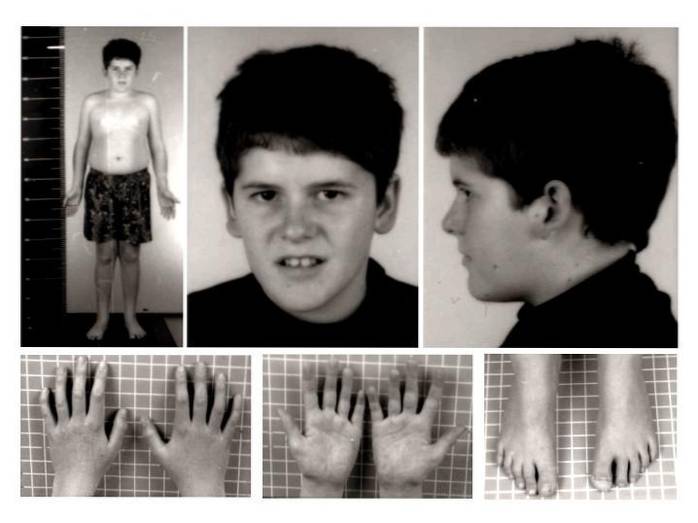
De mest karakteristiska fysiska tecknen och symtomen inkluderar störningar som; hypotoni, muskuloskeletala missbildningar eller missbildningar, minskad eller låg vikt och höjd, överdriven aptit, fetma, hypogonadism, sömnstörningar, andningsstörningar, atypiska lättdrag, förändring i regleringen av kroppstemperaturen, bland andra.
Förekomst eller utveckling av minskad muskeltonus. Den muskulösa slappheten i denna patologi accentueras särskilt i nacken och bagageutrymmet, särskilt i det nyfödda stadiet och de första månaderna av livet. Således, med biologisk utveckling, tenderar muskeltonus att förbättras.
I detta fall är det vanligt att observera utvecklingen av skolios eller avvikelse i ryggraden, dålig justering av underbenen (genu valgus) eller närvaron av platta fötter..
Dessutom kan andra typer av medfödda anomalier observeras, såsom minskning av storlek på fötter och händer, höftdysplasi, närvaro av sex fingrar, bland andra..
Särskilt vid tidpunkten för födseln är både det drabbade barnets längd och vikt lägre än förväntat för deras utveckling och kön. Trots det faktum att standardvärden kan uppnås i vuxen ålder tenderar den långsamma tillväxttakten att förändra vuxnas värden på höjd och vikt.
Det är vanligt att observera en omättlig aptit hos personer med Prader-Willis syndrom, som kännetecknas av en besatthet eller fixering av mat. På grund av intaget av stora mängder mat tenderar de drabbade att utveckla fetma och andra associerade medicinska komplikationer, såsom typ II-diabetes mellitus.
Förekomsten av könsförändringar är också frekvent. Specifikt är hypogonadism eller partiell utveckling av de yttre könsorganen mycket vanligt. I de flesta fall når inte pubertetsutvecklingen de slutliga eller vuxna stadierna.
Snarkning, ökad frekvens eller andningsstopp uppträder ofta återkommande under sömnfaser. De drabbade tenderar således att presentera olika förändringar relaterade till fragmentering, sömnfördröjning eller närvaron av periodiska uppvaknande.
Muskuloskeletala abnormiteter och missbildningar kan också påverka kraniofaciala egenskaper. Det är möjligt att observera en smal skalle, ögonskel, dåligt pigmenterad hud och hår, liten mun och tunna läppar, tandmissbildning etc..
Personer som drabbas av Prader-Willis syndrom har vanligtvis problem relaterade till reglering av kroppstemperatur, och ett annat viktigt resultat är hög motståndskraft mot smärta.
På grund av närvaron av muskuloskeletala missbildningar och minskad muskeltonus kommer psykomotorisk utveckling att gå långsammare, vilket påverkar alla områden.
De drabbade uppvisar vanligtvis seriens svårigheter att utföra någon typ av aktivitet som kräver en eller flera motoriska körningar.
När det gäller kognitiva begränsningar har de flesta av de drabbade en mild eller måttlig intellektuell funktionsnedsättning.
Utöver detta presenterar de vanligtvis vissa specifika områden som är mer påverkade, såsom sekventiell bearbetning av information, nyligen eller korttidsminne, lösning av aritmetiska problem, auditiv bearbetning av verbal information, förändring av uppmärksamhet och koncentration och närvaro av kognitiv stelhet.
Å andra sidan är språk ett annat område som påverkas signifikant hos individer med Prader-Willis syndrom. Förseningar i förvärv av fonologiska färdigheter, dålig ordförråd, förändring av bland annat grammatisk konstruktion observeras vanligtvis..
Beteendeproblem och förändringar är en annan av de typiska fynd som kan observeras i Prader-Willis syndrom, de varierar normalt beroende på ålder eller mognadsstadium där den drabbade personen är, men några av de vanligaste beteendemässiga egenskaperna är:
Olika aktuella undersökningar har påpekat att beteendeförändringar tenderar att öka med åldern och därför tenderar de att förvärras och påverkar sociala, familje- och emotionella områden på ett allmänt sätt.
Som vi har påpekat i flera avsnitt ovan har Prader-Willis syndrom ett genetiskt ursprung.
Även om det för närvarande är en stor kontrovers om de specifika gener som är ansvariga för denna patologi, visar all data att den etiologiska förändringen ligger på kromosom 15.
Under den genetiska studien av denna patologi har det funnits flera bidrag. Burtler och Palmer (1838) upptäckte förekomsten av avvikelser i den långa armen av kromosom 15 från faderns förälder, medan Nicholls (1989) observerade att i andra fall var sjukdomen relaterad till kromosomala förändringar från modern (Rosell-Raga, 2003).
Bortsett från detta är den mest accepterade teorin om ursprunget till denna patologi förlust eller inaktivering av olika gener för faderns uttryck som ligger i 15q11-13-regionen i kromosom 15.
Diagnosen av Prader-Willis syndrom har två grundläggande komponenter, analys av kliniska fynd och genetisk testning..
När det gäller upptäckt av indikatortecken och symtom, både hos spädbarn och hos äldre barn, är det viktigt att utföra en detaljerad, individuell och familjehistoria. På samma sätt är det också viktigt att utföra en fysisk och neurologisk undersökning..
Om det finns en diagnostisk misstanke baserad på dessa procedurer kommer det att vara nödvändigt att ordinera olika kompletterande tester för att bestämma förekomsten av genetiska förändringar och abnormiteter..
Specifikt diagnostiseras cirka 90% av fallen definitivt genom DNA-metyleringstester och andra ytterligare tester..
Dessutom är det också möjligt att ställa en prenatal diagnos av detta medicinska tillstånd, huvudsakligen i familjer med tidigare Prader-Willis syndrom..
Specifikt möjliggör fostervattentestet extrahering av embryoprover för utförandet av relevanta genetiska tester.
Det finns för närvarande inget botemedel mot Prader-Willis syndrom. Liksom vid andra sällsynta sjukdomar är behandlingar begränsade till symptomkontroll och förbättring av drabbade personers livskvalitet.
En av de grundläggande aspekterna är emellertid näring och kostkontroll, eftersom fetma är den främsta orsaken till sjuklighet och dödlighet i denna patologi..
Å andra sidan kommer närvaron av kognitiva och beteendemässiga förändringar att kräva intervention av specialister både i kognitiv rehabilitering och vid hantering av beteendestörning.

Ingen har kommenterat den här artikeln än.