De Duchennes muskeldystrofi (DMD) är en neuromuskulär sjukdom som kännetecknas av närvaron av signifikant muskelsvaghet och en generaliserad och progressiv utveckling (Världshälsoorganisationen, 2012).
Det är den vanligaste typen av muskeldystrofi hos människor (López-Hernández, 2009) och drabbar 1 av 3 500 barn i världen (Duchenne Parent Project, 2012). För det mesta drabbar sjukdomen män i de tidiga stadierna av livet (Världshälsoorganisationen, 2012).
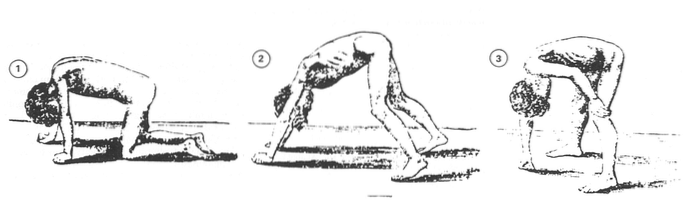
Det finns olika typer av muskeldystrofi. Symtom börjar vanligtvis under barndomen. Svaghet och förlust av muskelmassa orsakar allvarliga svårigheter att förvärva eller upprätthålla förmågan att gå, andas och / eller svälja (Mayo Clinic, 2013).
Neuromuskulära effekter ger en kronisk prognos. I de flesta fall dör personer med Duchennes muskeldystrofi i ung vuxen ålder på grund av utvecklingen av sekundära patologier som hjärtsvikt eller kardiomyopatier (Världshälsoorganisationen, 2012).
Artikelindex
Duchennes muskeldystrofi är en sjukdom som drabbar individen genom progressiv muskelsvaghet och degeneration (Muscular Dystrophy Association, 2016).
På grund av en genetisk mutation kommer frånvaron av ett specifikt protein hos personer med Duchennes muskeldystrofi att orsaka förlust av muskelfunktionalitet..
Vanligtvis uppträder symtomen i nedre extremiteterna och sprider sig till resten av områdena.
Världshälsoorganisationen (2012) indikerar att förekomsten av Duchennes muskeldystrofi uppskattas till ungefär 1 fall per 3300 invånare.
Specifikt visar vissa undersökningar att denna sjukdom drabbar 1 av 3 500 manliga barn födda levande (López-Hernández, 2009).
För USA är det inte säkert känt hur många människor i alla åldersgrupper som lider av denna sjukdom. Viss forskning har uppskattat att en av 5600-7 770 manliga vuxna i åldrarna 5 till 24 har en diagnos av Duchenne eller Becker muskeldystrofi (Centers for Disease Control and Prevention, 2015).
Det mest karakteristiska för störningarna som hör till gruppen av muskeldystrofi är muskelsvaghet; Beroende på typ kan dock specifika symtom förekomma som varierar beroende på åldersstart och de påverkade muskelgrupperna (Mayo Clinic, 2013).
Normalt är utvecklingen av Duchnne muskeldystrofi ganska förutsägbar. Föräldrar kan observera några ganska signifikanta tecken, såsom svårigheter eller oförmåga att lära sig gå eller onormal ökning av vadmusklerna (pseudohypertrofi) (Duchenne Parent Project, 2012).
Några av de mest karakteristiska symtomen och tecknen på Duchennes muskeldystrofi som uppträder tidigt i ett barns liv är (Mayo Clinic, 2013):
På samma sätt lyfter Duchenne Parent Projet Association (2012) fram de vanligaste symtomen och kliniska manifestationerna:
Alla muskelsymptom börjar med svaghet i bäckenbältets muskler, kalvar och olika gångstörningar som är signifikanta före 5 års ålder (López-Hernández, 2009).
I förskolan kan barn med Duchennes muskeldystrofi falla ofta eller ha svårt att gå, klättra steg och / eller springa (Duchenne Parent Project, 2012).
När sjukdomen fortskrider, i skolåldern, är det mycket troligt att barn bara använder fotspetsarna för att gå. Vi kommer att kunna observera en rullande och osäker marsch som kan orsaka många fall. De använder vanligtvis några strategier för att bibehålla sin balans, som att trycka tillbaka axlarna eller hålla fast i sin egen kropp (Duchenne Parent Project, 2012).
Cirka 9 år kan de flesta med denna sjukdom inte gå, på grund av detta börjar de utveckla många muskuloskeletala deformiteter - skolios, kontrakturer etc. - (López-Hernández, 2009).
I tonåren kommer de att presentera betydande svårigheter att effektivt utföra aktiviteter relaterade till användningen av övre extremiteter, ben eller bagage. I detta skede kommer de att behöva mekaniskt stöd och hjälp (Duchenne Parent Project, 2012).
Muskeldegeneration och svaghet fortsätter att gå fram tills de når musklerna som är ansvariga för andnings- och hjärtfunktionen (López-Hernández, 2009). På grund av allt detta äventyras patientens överlevnad allvarligt och orsakar död i de flesta fall..
Olika gener har identifierats som är involverade i produktionen av proteiner som är ansvariga för att skydda muskelfibrer mot eventuell skada och skada (Mayo Clinic, 2013).
Specifikt inträffar varje typ av muskeldystrofi som en följd av en viss genetisk mutation. Några av dessa mutationer ärvs; i de flesta fall uppträder de emellertid spontant under graviditeten (Mayo Clinic, 2013).
När det gäller Duchennes muskeldystrofi identifierade forskarna en specifik gen lokaliserad på X-kromosomen som skulle kunna presentera den mutation som är ansvarig för denna patologi (Muscular Dystrophy Association, 2016).
Således identifierades proteinet associerat med denna gen 1987., dystrofin. Således innebär bristen eller frånvaron av detta protein att musklerna är ömtåliga och lätt skadas (Muscular Dystrophy Association, 2016).
Dessutom har ett recessivt arvsmönster kopplat till X-kromosomen identifierats, med bäraren som moderen (Muscular Dystrophy Association, 2016). På grund av detta faktum är denna typ av sjukdom vanligare hos män än hos kvinnor..
Män har en XY-kromosomkomposition, medan kvinnor är XX. Därför, om en X-kromosom har en mutation i DMD-genen, kommer den att drabbas av Duchennes muskeldystrofi på grund av frånvaron av dystrofinproduktion (National Human Genome Research Institute, 2013).
När det gäller kvinnor som har två X-kromosomer och därmed två kopior av DMD-genen, kommer den andra att fortsätta att producera dystrofin och därmed upprätthålla muskelneuroskydd (National Human Genome Research Institute, 2013). ).
I denna typ av patologier kan olika ingrepp utföras för att bestämma diagnosen (National Human Genome Research Institute, 2013).
Den kliniska diagnosen kan redan ställas när ett barn börjar utveckla progressiv muskelsvaghet. Redan vid 5 års ålder finns det uppenbara symtom. Om ett tidigt ingripande inte genomförs kommer barn att ha funktionsberoende före 13 års ålder (National Human Genome Research Institute, 2013).
Förutom observation och klinisk undersökning kan några av följande tekniker användas för att identifiera närvaron av Duchennes muskeldystrofi (Mayo Clinic, 2013):
För närvarande har ett botemedel mot Duchennes muskeldystrofi inte identifierats (Duchenne Parent Project, 2012).
Trots detta används olika behandlingar som har visat sig vara effektiva både för att minska symtomen och för att förbättra livskvaliteten för människor som lider av denna typ av patologi (Duchenne Parent Project, 2012).
På grund av den kliniska utvecklingen och den stora variationen av symtom kommer denna sjukdom att kräva ett tvärvetenskapligt och omfattande ingrepp utfört av en mängd olika specialister: barnläkare, sjukgymnast, neurolog, neuropsykolog, arbetsterapeut, logoped, nutritionist, endokrinolog, genetiker, bland annat kardiolog, lungläkare, ortoped, rehabilitator och kirurg (Duchenne Parent Project, 2012).
I många fall kan specialister rekommendera farmakologiska ingrepp (Mayo Clinic, 2013):
Läkemedel är inte bara användbara för intervention i Duchennes muskeldystrofi, det finns både terapeutiska ingrepp och vårdmetoder som kan förbättra livskvaliteten för dessa människor (Mayo Clinic, 2013).
Några fördelaktiga insatser är (Duchenne Parent Project, 2012):
Fram till för relativt få år sedan överlevde personer med Duchennes muskeldystrofi inte mycket längre efter att de uppnått tonåren (Muscular Dystrophy Association, 2016).
De stora framstegen inom medicinsk, teknisk och genetisk forskning har lyckats både sakta sjukdomsprogressionen och ge en betydande ökning av livskvaliteten för individer som lider av den (Muscular Dystrophy Association, 2016). På detta sätt är hjärt- och andningsvård avgörande för att bevara vitala funktioner (Muscular Distrophy Association, 2016).
I många fall är de kapabla att nå de post-tonåriga stadierna. Fler och fler fall av Duchennes muskeldystrofi beskrivs hos vuxna i 30-talet, inklusive personer som överlever till 40- och 50-talet (Muscular Dystrophy Associatin, 2016).
För närvarande är kliniska prövningar och forskning inriktade på utveckling av genterapier som modifierar mutationer och brister i dystrofinproduktionen (Muscular Dystrophy Association, 2016).
Några av de mest undersökta mekanismerna är (López-Hernández, 2009):
Duchennes muskeldystrofi är en allvarligt inaktiverande sjukdom hos både barn och unga vuxna med en förödande prognos..
Trots det faktum att klinisk och experimentell forskning har gjort viktiga framsteg i behandlingen av symtom, finns det fortfarande inget botemedel mot denna typ av patologi..
En grundlig förståelse av den biologiska och genetiska grunden är nödvändig för att hitta en botande behandling för Duchennes muskeldystrofi..


Ingen har kommenterat den här artikeln än.